26 July 2013
Aphaenogaster, RNAseq, transcriptome, FLASH, digital normalization, and diginorm
Next post Previous postRNAseq read filtering prior to assembly
ApTranscriptome
Confirmed yesterday that running velvet (or Trinity) for transcriptome assembly will more RAM than available on antlab. Initiated process to get server time on mason (512GB nodes!).
While waiting, started looking into alternate methods and assemblers. Two promising approaches to reduce data:
- merging paired-reads (FLASH)
- digital normalization
Merge paired reads
I thought that FLASH would not work to merge reads because insert size is 200 bp and reads are only 100 bp, so after filtering there may not be enough overlap. Example uses 175 bp inserts. However, as read length is normally-distributed (see FLASH paper) many inserts will be less than 200 bp and may merge so worth a try to reduce the number of reads in data set. Unmerged reads will simply be returned as is.
Awesome! For first sample, 67% of reads were merged after running FLASH:
| Sample | Total Reads | Merged | Unmerged |
|---|---|---|---|
| A22-00-R1 | 16,351,272 | 11,060,076 | 5,291,196 |
And a histogram of the distribution of length of extended fragments.
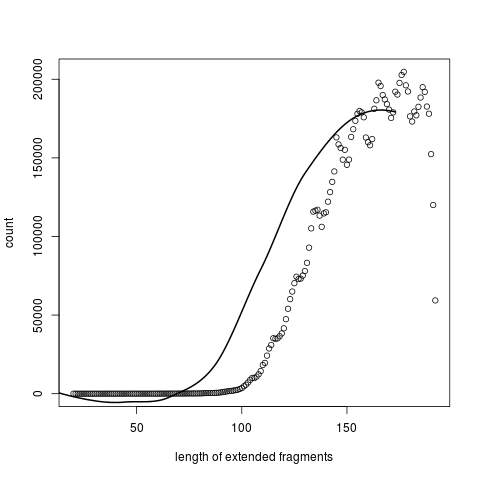
Note that though the library was size selected to 200bp, the extended fragments have a mode length around 160, probably due to some trimming, but also must be enough overlap to allow combining.
Ran for all samples in ~ 1.5 hrs. 64-70% of reads merged - huge reduction in total file size for assembly!
Digital normalization
After merging reads, perform digital normalization to discard redundant data (lower memory for assembly) and error removal.
Following the diginorm tutoral guidelines. Had to make changes as I am not doing this on EC2.
- Set PYTHONPATH to khmer location. This website helped understand PYTHONPATH
export PYTHONPATH=/opt/software/khmer/python- Setting -N and -x parameters following guidelines
- “parameters specify the maximum memory usage of the primary data structure in khmer, which is basically N big hash tables of size x. The product of the number of hash tables and the size of the hash tables specifies the total amount of memory used.”
- “absolute safest thing to do is to specify as much memory as is available”
-n 4 -x 4e9==> 1.6e10 ==> 16GB. ‘Rules of thumb’ suggested this adequate for up to a billion mRNAseq reads- went with
-n 4 -x 2e9as each of my samples has far fewer than a billion reads
- Set -C (coverage threshold) to 20 as this was the standard used in the paper
- Set kmer size to 21 as this was used for yeast mRNA in paper
- According to this exchange on khmer list, can perform normalization on each sample individually and then a second pass on all combined with very similar results to a single pass. Documented in the section on ‘Iterative and independent normalization’ ~ normalize-by-median [ … ] file1.fa normalize-by-median [ … ] file2.fa normalize-by-median [ … ] file3.fa normalize-by-median [ … ] file1.fa.keep file2.fa.keep file3.fa.keep ~
- According to this exchange on khmer list, the
-poption keeps or rejects both reads based on whether either one is novel; though nobody has really explored the effects of this on assembly.
Set up script to do this and started running…
Todo
- Run FastQC on merged reads
