de novo gene expression mapping works!
Gene expression counts
With BWA, have actual read counts, but need normalized counts - Reads per Kilobase per Million mapped reads (RPKM) typically reported but recently shown that this is problematic for comparison among samples so Transcripts per Million (TPM) is endorsed (Wagner et al. 2012). Bizarrely, couldn’t find any scripts to do this from BWA and the original paper not clear on how to do so. Fortunately found great blog post with a nice worked example that I was able to recreate.
Made my example available as a gist
Useful presentation on TPM here
Success!!! Working with TPM instead of raw gene counts results in a strong correlation (r = 0.90) between known expression and TPM from mapping reads to Oases transcriptome. Resolves issue from first round of gene expression simulation.
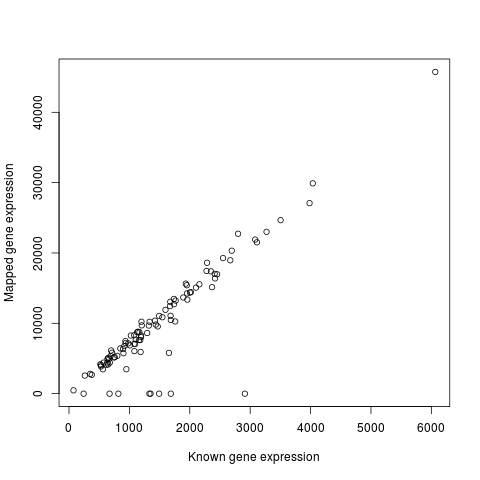
Now that I know it works and have a functioning pipeline, proceed to full analysis!
BWA mapping
When I map reads to the known transcripts, results are as such
| Locus | Start | Stop | Count |
|---|---|---|---|
| ENA|AAA02840|AAA02840.1 | 0 | 1092 | 18584 |
| ENA|AAA03449|AAA03449.1 | 0 | 765 | 7458 |
| ENA|AAA16569|AAA16569.1 | 0 | 504 | 5984 |
| ENA|AAA16570|AAA16570.1 | 0 | 522 | 7236 |
| ENA|AAA16571|AAA16571.1 | 0 | 558 | 14094 |
where the first column is the gene, second and third columns are start and stop locations where reads were aligned to, and fourth column is the number of reads mapping to that transcript.
However, when I map reads to Oases assembled transcripts, I get NO mapped reads
| Locus | Start | Stop | Count |
|---|---|---|---|
| Locus_1_Transcript_1/10_Confidence_0.450_Length_1487 | 0 | 1487 | 0 |
| Locus_1_Transcript_2/10_Confidence_0.325_Length_1543 | 0 | 1543 | 0 |
| Locus_1_Transcript_3/10_Confidence_0.550_Length_1398 | 0 | 1398 | 0 |
| Locus_1_Transcript_4/10_Confidence_0.525_Length_1705 | 0 | 1705 | 0 |
| Locus_1_Transcript_5/10_Confidence_0.525_Length_1401 | 0 | 1401 | 0 |
| Locus_1_Transcript_6/10_Confidence_0.450_Length_1669 | 0 | 1669 | 0 |
Odd. Maybe because of multiple isoforms in Oases assembly..though I thought it tried to take them out by ‘popping bubbles’
