16 August 2013
Aphaenogaster, RNAseq, transcriptome, simulation, fastq, and git
Next post Previous postWeek's notes August 16
ApTranscriptome
Set up to run transcriptome assembly on mason cluster. Access by ssh username@login.xsede.org and then running gsissh project-username@mason.iu.xsede.org. Transferred Illumina data to mason data capacitor /N/dc/scratch/tg-johnsg
Note that files must be accessed every 60 days or will be purged.
Seems advisable to set up checksum to check data integrity
Transfer completed in ~8 hours:
sent 927 bytes received 57941491018 bytes 2076272.26 bytes/sec
total size is 189387908128 speedup is 3.27in silico mRNA spike-in to validate transcriptome assembly.
in progress. status to date
Get sample of sequences in FASTA format to simulate reads. These are simulated ‘known’ mRNA fragments floating around in my eppendorf after RNA extraction.
cf10104_cra10 CCGATACGACATGTCAG…
Using the
seltool inrlsimassign an expression-level to each fasta sequence. Default is to use a mixture of gamma distributions with two components of mean 5,000 and 10,000. The output of this is simply the same fasta sequences but annotated with expression levels by a $XXXX added to the fasta title, where XXXX is the expression level. For example
Output looks like
>cf10104_cra10$51373
CCGATACGACATGTCAG...For my data, a full lane for each colony ~ 160 million reads. With 17k transcripts expected based on ant genomes and average length of 1k bp per transcript, average coverage should be (160e6*200)/(17e3*1e3) = 1882x. Of course, this will be highly variable due to differences in expression so re-sample expression levels as a gamma distribution centered on this value. For simulated data, don’t need to guess ‘truth’ but should simulate some genes with low expression (~10), some around median (~1500) and some with high expression (~10000). Can then evaluate accuracy of assembly for genes with low, median and high expression levels.
sel requires a gamma distribution for expression levels. Following the specification on p11 of the rlsim manual, can specify an equal mix of these distributions by : 1:g:(10, 1) + 1:g:(1500, 3) + 1:g:(10000, 10, ). Based on mean, selected shape parameters by experimentin R. Plot of simulated distribution looks like this 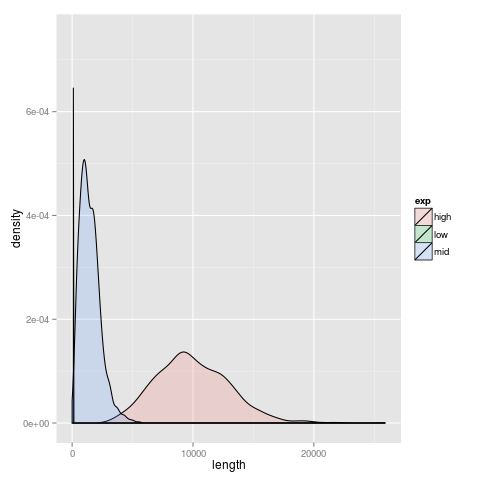 .
.
Full command is:
sel -d "1:g:(10, 1) + 1:g:(500, 3) + 1:g:(1000, 10)" ../data/ENA-arabidopsis-random-mRNA.fasta > simulated.fastaSimulate fragments with default parameters using
rlsim. Hmmm…how many fragments to simulate? Seems thatefftestis made (partly) for this purpose, but requires paired-end reads mapped to a transcriptome…which I don’t have. Could get this iteratively, by making transcriptome and mapping reads, then using this to make simulated data, then repeating transcriptome assemly including simulated data and evaluate.rlsim -n 1000 simulated.fasta > Cf-frags.fasta
Generate paired-end sequencing reads using simNGS
cat Cf-frags.fasta | simNGS -p paired -o fastq -O reads /opt/software/simNGS/data/s_3_4x.runfile
After running through simNGS the number of reads is equivalent to the number of fragments sampled…which makes sense. So, to decide number of fragments I need to decide how many reads I want. For a transcript of 1000bp at the median coverage of 1800x I would want (1e3 * 1.8e3) / (2 * 100) = 900 fragments.
Clearly, I need to evalute if this simulation business works at all given how I’ve set it up. So, set up an independent run of assembly using only simulated data. If this works…proceed.
Downloaded first 10,000 fasta mRNA transcripts from European Nucleotide Archive as yesterday.
ApAdaPt
Started knockdown time assays Wednesday 2013-08-13. Will detail protocol soon…. Takes ~4.5 hrs to knockdown assay for a colony with 40 ants. Some concerns about the precision of the assay as it can be difficult to definitively determine when ant is ‘knocked’ down. Decided to record to closest half-minute as this is the accuracy that I feel I have.
Status
| Date | Colony completed |
|---|---|
| 13 Aug | AA05 |
| 13 Aug | AA11 |
| 14 Aug | AA10 |
| 14 Aug | AA02 |
| 15 Aug | AA04 |
| 16 Aug | AA06 |
| 16 Aug | AA03 |
Computing
Cool trick for counting reads in fastq file by command line:
echo wc -l reads.fq | cut -f 1 -d ' ' / 4 | bc
git
push new local branch to remote repository with
git push -u origin newbranchMore git brilliance: use git submodules to include other git repositories within repo. Basic workflow explained on this biostars post.
